You must demonstrate that you can detect the transgene to ensure that 1) you can genotype the founder mice and 2) to ensure that you will be able to maintain the lines without loss.
Our recommendation is that you use Southern blot analysis to identify founder animals. Southern blot analysis is preferred for the identification of founder animals because it has a low false positive rate. PCR can be used on the offspring of the founders, and the PCR assay can be validated against the Southern blot results of the founders. Whenever PCR is used for genotyping, we recommend that positive and negative controls always be included (see below).
A restriction endonuclease recognition site which occurs only once in the transgene should be used to cut the genomic DNA, and this sequence should not contain the sequence 5'-CG-3': this sequence is usually methylated and resistant to digestion in mammalian genomic DNA. Because the construct usually integrates in multiple head-to-tail copies at a single site, cutting with an enzyme with one site in the transgene will result in one fragment the same size as the transgene, and in the end fragments of the transgene array where they meet with genomic DNA. Because the arrayed transgene is often in tens to hundreds of copies, the band the size of the transgene will result in a much stronger band than the junction fragments.
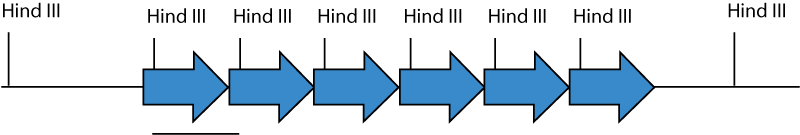
Each arrow represents a single copy of the transgene. The transgene is inserted in multiple head-to-tail copies at one place in the genomic DNA. Hind III cuts once in the transgene. Genomic DNA cut with Hind III releases 5 copies of a fragment the size of the transgene, and two fragments which are junction fragments with genomic DNA.
Because the transgene arrays made from plasmid transgenes can undergo DNA methylation and irreversible extinction of expression, you might consider developing an assay for the expression of the transgene. Ideally, an expression assay is used for monitoring transmission of the transgene. Extinction of transgene expression is irreversible, and occurs both in the germline, and in the soma. Silencing can be mosaic in the soma, and can increase with age. BAC transgenes, gene traps, and knockouts/knockins and mice generated by homologous recombination typically do not undergo methylation-based silencing. Factors contributing to plasmid transgene shutdown include the repetitive nature of the transgene arrays, CpG content, effects from the integration site and the strain of mice (see references at the bottom of the page)
Designing and Validating PCR Assays
PCR false positives arise from contamination and low primer specificity. The fragment amplified by the primers should be specific to the transgene--for example, primers to beta-gal alone are not a good idea since a very large number of plasmids and mice carry the same sequence. The best primer pairs bridge between two portions uniquely combined in the transgene, for example between the promoter and coding sequences of a transgene.
It may take several tries to find primer pairs which work well in the context of mouse genomic DNA. Use Primer3 to pick several primer pairs.
Validation of PCR Assays
To validate a PCR assay, a negative control of wild type mouse genomic DNA and positive controls of wild type mouse genomic DNA spiked with transgene at 1, 0.1, 0.01, 0.001 and copies per diploid genome should be performed. This series will demonstrate the sensitivity of the assay reveal background problems--a point of no detection should be reached. It is not adequate to test the PCR assay on dilutions of construct DNA in the absence of genomic DNA. Markers should be run on the same gel in order to determine if the amplified product is of the correct size. A positive control for the reaction components and genomic DNA should be performed. The primers below reliably amplify a 300 bp fragment from the mouse Notch1 gene using these PCR reaction conditions.
N1.2: 5' GTG GTC TAG GAT GCT TGG GTC TAG 3'
The number of cycles should be no more than 30-40, and should be performed on 0.2, 2 and 20 ng of genomic DNA. If the PCR reaction shows a smear in the lane of the gel, lower the amount of input DNA.
Calculate Transgene Mass To Spike One Copy Per Diploid Genome
The University of Michigan Transgenic Animal Core and other cores provide calculations based on one copy per haploid genome, and thus arrive at values twice as high.
Controls for PCR Genotyping in the Propagation of Transgenic Mice
Because of the sensitivity of PCR, cross-contamination between mice can occur by carryover on the punch or forceps. Clidox will interfere with PCR reactions, so rinse the punch, forceps or other tools with 70% ethanol between samples. The punch and forceps need to be inspected between mice, and control samples of sequential known transgenic followed by known wild type mice should be taken at every tissue collection session to monitor cross-contamination. Markers should be run on every gel, to ensure that the product is of the correct size, and only samples with robust bands should be scored as transgene-positive.
Troubleshooting Southern Blots
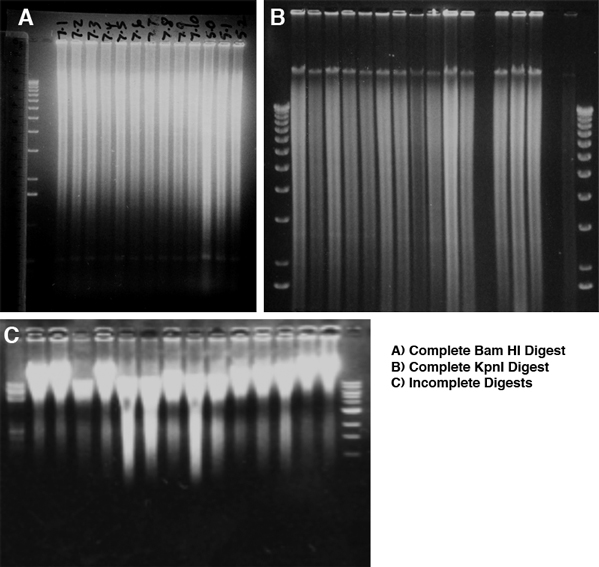
Genomic DNA is viscous and requires more restriction enzyme per unit DNA and more time to cut than plasmid DNA. Use high concentration enzyme (50 U/microliter), but the total volume of restriction enzyme added must be 10% or less of the complete reaction volume. Incubate the digestion reaction for 12-24 hours. Add half of the enzyme, incubate for a few hours to overnight, then mix gently and add the remainder of the enzyme and incubate for the remainder. As a first approximate measure of digestion, the solution should no longer be viscous--this however does not ensure that digestion is complete. To determine if the cut is complete, run and stain the gel with ethidium bromide and photograph it before attempting to transfer. The stained DNA should be distributed in a graded smear, with the highest intensity at the top, around 20 kb, with a linear rather than logarithmic decrease in intensity. With many restriction enzymes there will be band(s) in the smear representing the single unit from arrayed repetitive DNA. Examples of complete and incomplete digests are shown here. With successful digestion, all lanes will have identical smears and faint bands of similar intensity. With partial digests, the degree of digestion will vary, and the less cut samples will have more DNA at the top, and the intensity of the smear will decrease sharply from top to bottom of the gel. Uncut DNA will run at about 40 kb, the size it was physically sheared to during purification. Don't blot and hybridize to partial digests -- start new cuts rather than continuing if the DNA is not completely cut, since partial digests are invariably uninterpretable.
{kind=link}
If the ethidium-stained gel indicated a complete cut, then we can move on to problems with signal and mysterious bands.
You should be able to detect single copy DNA in 2 micrograms of genomic DNA with an O/N to 2 day exposure with a probe 1 kb in length which was labeled by random prime labelling with high specific activity 32P-dCTP.
No signal: probe not labeled; probe wasn't denatured; fragment too big to transfer efficiently to membrane.
A consistent band in all samples, including DNA from uninjected zygotes: cross-hybridization to a related sequence--as long as it isn't similar in size to the expected fragment, this should not be a problem.
In founder animal samples, smaller than expected bands: 'star' activity (relaxed or altered specificity of the restriction enzyme due to lower ionic strength)--check the salt concentration and that enzyme amount is 10% or less of the reaction volume.
In founders, bands of variable unexpected larger sizes: partial digestion; the junction fragments of the transgene array.
References and Links
Silencing of Plasmid Transgenes:
- Nucleic Acids Research, 1996, 24: 1465-1471 (.pdf)
- BioEssays 1998, 20: 532-535 (.pdf)
- Transgenic Research 2000, 9: 237-239 (.pdf)
- Experimental Cell Research 2002, 274: 1-8 (.pdf)
- Genome Biology 2003, 4: R53 (.pdf)
BAC Transgenics:
Protocols
- Breeding and Maintaining Mice
- Quick Ear Punch protocol for marking mice and collecting DNA for PCR
- DNA Isolation for Southern Blot
External Links
- Southern Blot Rationale and Protocol (NEMC-TUSM Transgenic Core Facility)
- Preparation of DNA from Tail Samples for Southern Blot Analysis (NEMC-TUSM Transgenic Core Facility)